
This is the fourth part in a short series of blog posts about quantum Monte Carlo (QMC). The series is derived from an introductory lecture I gave on the subject at the University of Guelph.
Part 1 – calculating Pi with Monte Carlo
Part 2 – Galton’s peg board and the central limit theorem
Part 3 – Markov Chain Monte Carlo
Introduction to QMC – Part 4: High dimensional calculations with VMC
In the previous post, Markov Chains were introduced along with the Metropolis algorithm. We then looked at a Markov Chain Monte Carlo (MCMC) Python script for sampling probability distributions. Today we’ll use a modified version of that script to perform calculations inspired by quantum mechanics. This will involve sampling high dimensional probability distributions for systems with multiple particles.
We are heading into complicated territory, so I set up the first section of this post in question-answer format.
Why is it called VMC and not MCMC?
As far as I can tell, variational Monte Carlo (VMC) is the same as MCMC. The name was probably inspired by the variational theorem from quantum mechanics, but we’ll get to this later. For now let’s simply pose the problem:
Given a probability distribution 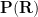
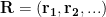


Why do we need to worry about probability distributions? Because in the quantum world things to not always have well defined positions, but instead only have well defined probabilities of being in specific positions. Hence the existence of probability distributions.
What function are we calculating?
We’ll calculate the average value of the local energy 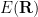
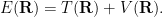
It’s not unreasonable to plug in some 
Kinetic term T
The kinetic energy will depend on the second derivative of the wave function, which is relatively difficult and computationally time consuming to calculate. To avoid dealing with this beast we’ll use a made-up function
![T(\mathbf{R}) = \Big[\sum_i^{N/2}|\, x_i \cdot y_i \, |\Big] \cdot \Big[\sum_{j'}^{N/2} | \, x_{j'} \cdot y_{j'} \, |\Big].](https://s0.wp.com/latex.php?latex=T%28%5Cmathbf%7BR%7D%29+%3D+%5CBig%5B%5Csum_i%5E%7BN%2F2%7D%7C%5C%2C+x_i+%5Ccdot+y_i+%5C%2C+%7C%5CBig%5D+%5Ccdot+%5CBig%5B%5Csum_%7Bj%27%7D%5E%7BN%2F2%7D+%7C+%5C%2C+x_%7Bj%27%7D+%5Ccdot+y_%7Bj%27%7D+%5C%2C+%7C%5CBig%5D.&bg=ffffff&fg=000000&s=0&c=20201002)
It could be interpreted as the particles having more kinetic energy (i.e., larger 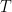
Potential term V
This is the cumulative potential energy of the particles, and unlike 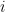
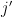

where 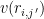
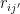
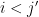
from scipy import stats
mu = 0.5
sig = 0.1
y = lambda r: -stats.norm.pdf((r-mu)/(sig))

We’ll focus on the case where 
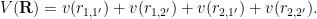
What probability distribution will we sample?
From elementary quantum mechanics, the probability density is given by the square of the wave function. So, for our many-body wave function 

The absolute value is meaningful when the wave function has imaginary components, which is not the case for us today. Our first 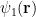

![\Psi_V(\mathbf{R}) = \sum_i^N \Big[\psi_1(\mathbf{r}_i)+\psi_2(\mathbf{r}_i)\Big].](https://s0.wp.com/latex.php?latex=%5CPsi_V%28%5Cmathbf%7BR%7D%29+%3D+%5Csum_i%5EN+%5CBig%5B%5Cpsi_1%28%5Cmathbf%7Br%7D_i%29%2B%5Cpsi_2%28%5Cmathbf%7Br%7D_i%29%5CBig%5D.+&bg=ffffff&fg=000000&s=0&c=20201002)
def prob_density(R, N):
''' The square of the many body wave function
Psi_V(R). '''
# e.g. for N=4:
# psi_v = psi(r_1) + psi(r_2) + psi(r_3) + psi(r_4)
psi_v = sum([psi_1(R[n][0], R[n][1]) for n in range(N)]) + \
sum([psi_2(R[n][0], R[n][1]) for n in range(N)])
# Setting anything outside the box equal to zero
# This will keep particles inside
for coordinate in R.ravel():
if abs(coordinate) >= 1:
psi_v = 0
return np.float64(psi_v**2)
Our configuration space is two dimensional (so we can visualize it nicely) and so particle positions consist of just 
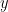
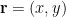
def psi_1(x, y):
''' A single-particle wave function. '''
g1 = lambda x, y: mlab.bivariate_normal(x, y, 0.2, 0.2, -0.25, -0.25, 0)
return g1(x, y)
def psi_2(x, y):
''' A single-particle wave function. '''
g2 = lambda x, y: -mlab.bivariate_normal(x, y, 0.2, 0.2, 0.25, 0.25, 0)
return g2(x, y)
The summation of

If we take this summation to be the wave function of a system with just one particle then the associated probability distribution for the particle location would look like:
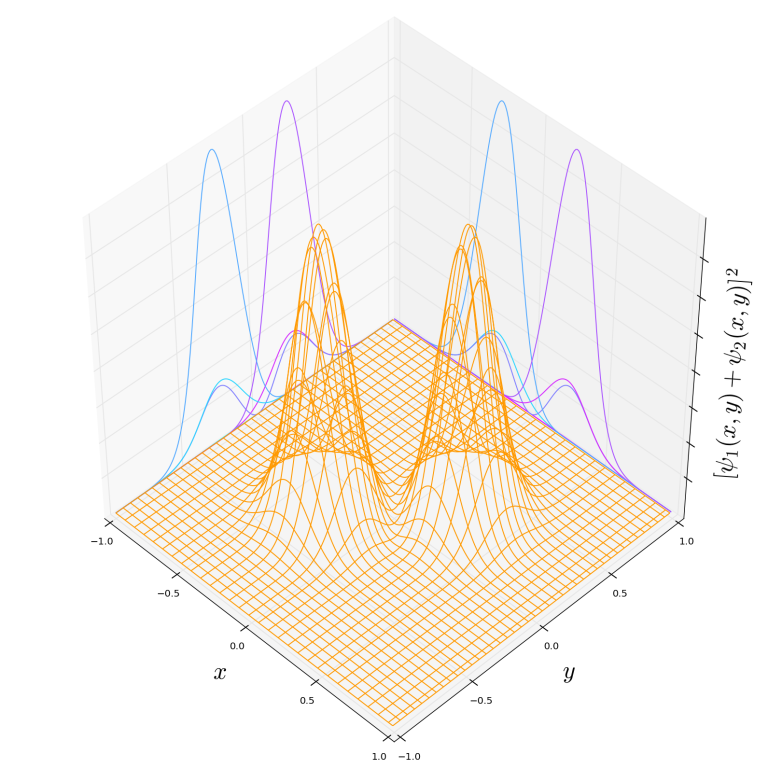
So what about a plot of the many-body wave function 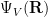

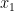
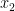

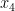


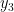
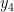
Calculating the average energy
We’ll focus on the
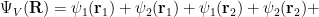
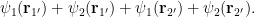
Running a quick simulation with 1000 walkers (i.e., 1000 samples per step) for 40 steps, the samples look like this:

The top left panel shows the initial state where walkers are distributed randomly. As the system equilibrates we see them drifting into areas where the probability density 
To calculate 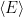

Each point is an average over all walkers at the given step. In this case we calculate an average energy of 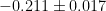

Running a calculation with 2000 walkers, we can see a dramatically reduced error:
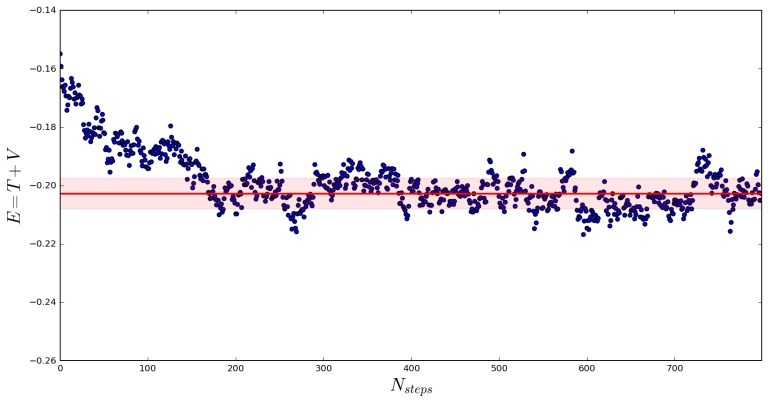
Now we calculate 
Equilibration times
In the calculations above, the system appeared to equilibrate almost immediately. This is because the initial configurations were randomly distributed about the box. If instead we force the particles to start near the corners of the box (far away from the areas where the probability distribution is large) we can clearly see

Variational Theorem
The usefulness of VMC for quantum mechanical problems has to do with the variational theorem. In words, this theorem says that the energy expectation value (denoted 

where 
If the name of the game is to find the ground state energy, which is often the case, then a good estimate can be achieved using VMC. A
Calculating average energy with a different many-body wave function
To show how changing
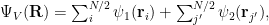
where the two species
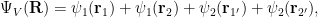
The new probability density will be:
def prob_density(R, N):
''' The square of the many body wave function
Psi_V(R). '''
psi_v = sum([psi_1(R[n][0], R[n][1]) for n in range(int(N/2))]) + \
sum([psi_2(R[n][0], R[n][1]) for n in range(int(N/2),N)])
# Setting anything outside the box equal to zero
# This will keep particles inside
for coordinate in R.ravel():
if abs(coordinate) >= 1:
psi_v = 0
return np.float64(psi_v**2)
This should have the effect of separating the two species of particles. Do you think
Plotting some of the samples, we can see how the system now equilibrates according to the new probability density:

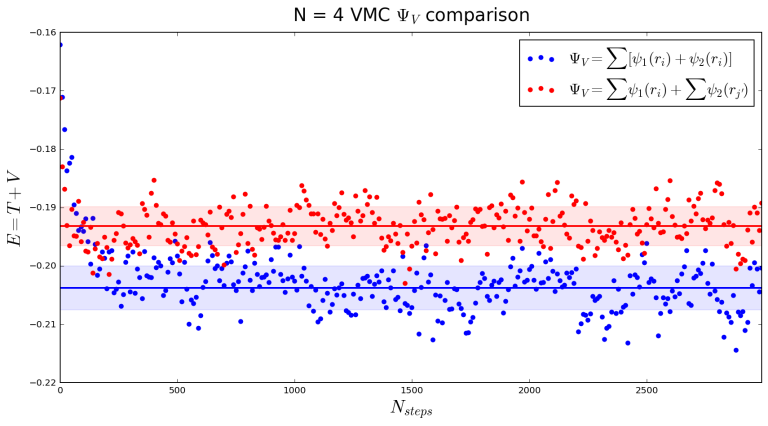
Comparing a calculation with the new
Thanks for reading! You can find the entire ipython notebook document here. If you would like to discuss any of the plots or have any questions or corrections, please write a comment. You are also welcome to email me at agalea91@gmail.com or tweet me @agalea91
[1] – For example we could have a system of cold atoms with two species that are identical except for the total spin. Where one species is spin-up and the other is spin-down.
[2] – I made the many-body wave function a sum of single-particle wavefunctions 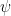
[3] – In practice, the error on Monte Carlo calculations is often given by the standard deviation divided by 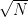


Hi, I think you may have a mistake, at the bottom of the part “WHAT FUNCTION ARE WE CALCULATING?” ,the formula V(R) should be V(R)=v(r1,1′)+v(r1,2′)+v(r2,2′)+v(2,1′), I don’t understand why you didn’t write the last term , or maybe I was wrong. So, if you know why, please help and explain to me. thanks.
LikeLike
You are correct, that term should have been included. I have made your change.
LikeLike
I also don’t understand why you write the many body wave function as the sum of single particle wave function , I think this is not correct.
LikeLike
What I have done should not be done in practice (and I regret doing it here). A common way to combine single particles wave functions is in a product (see my newly added point [2]). All I am doing is “guessing” a wave function (which is of course allowed) and performing VMC with that wavefunction. Then when I “guess” a different one we see a change in the VMC result.
Thanks so much for your comments!
LikeLike
I got it. Thank a lot.
LikeLike